 26-07-2019
26-07-2019 0 Просмотров
0 Просмотров 0 комментариев
0 комментариев 0 Рейтинг
0 Рейтинг
Аномалия Арнольда-Киари или как ее еще называют – мальформация – заболевание, знакомое медицине уже более сотни лет. Такой диагноз ставится при наличии грыжи в нижнем отделе мозга. При невнимательном к нему отношении недуг может иметь неприятные последствия.
Синдром Арнольда-Киари – что это?
Так называется врожденное заболевание, при котором наблюдается следующее: размеры ямки черепа, что сзади, и расположенных внутри нее структур не соответствуют друг другу. В результате мозжечковые миндалины и ствола мозга опускаются в большое отверстие в затылочной части черепа или защемляются. Синдром Арнольда-Киари диагностируется не очень часто. По статистике, его ставят не чаще, чем в 10 случаях на 100 тысяч населения. Средний возраст подверженных болезни – от 25 до 40 лет.
Аномалия Арнольда-Киари чревата нарушением оттока ликвора. Эта жидкость защищает мозг – как головной, так и спинной, – и если она не будет циркулировать корректно, может начаться блокада сигналов, которые мозг передает разным органам. В отдельных случаях происходит скопление ликвора, что тоже негативно сказывается на состоянии здоровья. При сильном давлении мозжечка на элементы нервной системы в позвоночнике иногда развивается сирингомиелия.
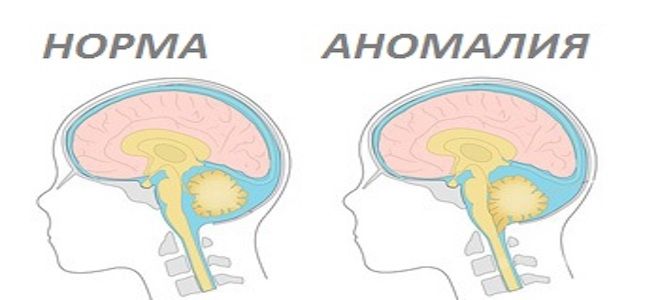
Аномалия Арнольда-Киари – степени
Определять их должен специалист. Сегодня медиками принято выделять две основные разновидности заболевания:
- аномалия Арнольда-Киари 1 степени – проблема, вызванная врожденными изменениями;
- 2 степень – недуг развивается на фоне травм, полученных человеком при рождении или в ходе жизненного процесса.
Аномалия Арнольда-Киари – типы
Выделяют 4 основных. В зависимости от того, какого типа аномалия Арнольда-Киари, изменяется и лечение, потому определить его очень важно. Итак, болезнь бывает:
- Аномалия Арнольда-Киари 1 типа. При этой форме недуга миндалины мозжечка опускаются не очень сильно.
- Для 2 типа характерны более грубые изменения, и заключаются они в смещении нижних отделов мозжечкового червя, 4 желудочка и продолговатого мозга.
- Аномалия 3 типа Арнольда-Киари встречается редко. При таком диагнозе задний мозг смещается в канал позвоночника.
- При 4 типе доктора диагностируют недоразвитие мозжечковой структуры.
Мальформация Арнольда-Киари 1 типа считается самой легкой и простой в лечении. Третий и четвертый тип заболевания – самые опасные. Как показывает практика, жить с такими формами болезни пациенты не могут. Кроме всего прочего, есть еще и аномалия типа «зеро». Ее ставят, если у пациентов уже развивается сирингомиелия, но миндалины мозжечка при этом опускаются не слишком сильно.
Синдром Арнольда-Киари – причины
Хоть болезнь и изучают тщательно многие специалисты, определить точно, почему она появляется пока что никто так и не смог. В теории, аномалия Арнольда-Киари 1 степени или 2 может развиваться из-за:
- генетических факторов;
- травм;
- гидродинамического удара ликвора.
Как показывает практика, синдром Арнольда-Киари у плода причины имеет самые вероятные. Располагающими факторами к развитию такой аномалии можно считать:
- злоупотребление матерью разными медицинскими препаратами;
- вирусные болезни во время беременности;
- курение и злоупотреблением алкоголем в период ожидания малыша.

В некоторых случаях болезнь становится следствием неправильного формирования костей внутри черепной коробки. Пациенты с приобретенной мальформацией страдают из-за травм, которые им нанесли во время родов. В процессе жизни получить такой диагноз также можно на фоне:
- уменьшения задней ямки черепа (когда мозжечку начинает не хватать места, он старается «выпихнуть» свою нижнюю часть туда, где больше свободного пространства);
- увеличения большого затылочного отверстия в размере.
Синдром Арнольда-Киари – симптомы
Они бывают общими или специфическими. Как правило, сначала появляются боли в затылке, которые у многих пациентов переходят и на область шеи. Во время приступов кашля или икоты неприятные ощущения становятся сильнее. Сопровождается все это исчезновением чувствительности кожи. На более сложных стадиях пациенты могут жаловаться на головокружение и проблемы со зрением – игнорировать такие признаки нельзя. Если они появляются, значит мальформация Арнольда-Киари прогрессирует.
К тревожным симптомам относят и такие:
- слабость в руках;
- покалывание в кистях;
- короткие остановки дыхания;
- нистагм;
- проблемы с мочеиспусканием;
- боль в голове;
- нарушения координации;
- тремор;
- ощущение постоянной усталости;
- рвота (которая никак не связана с употреблением пищи и начинается просто из-за повышения давления ликвора);
- понижение слуха.
Синдром Арнольда-Киари – лечение
Чтобы вылечить диагноз Арнольда-Киари, нужно хорошо изучить болезнь и особенности ее течения в конкретном организме. На выбор терапии влияет, тип, стадия, наличие или отсутствие осложнений. Лечение при синдроме Арнольда-Киари бывает консервативным или оперативным. На начальных этапах получится справиться и медикаментами (правда, нужно будет регулярно ходить на осмотры к специалистам). С болезнью же в запущенной форме приходится бороться только с помощью операции.
Аномалия Арнольда-Киари – клинические рекомендации
Если заболевание обнаруживается случайно – во время МРТ, например, – никакой терапии, как правило, не назначается. За пациентом просто устанавливается наблюдение, чтобы специалист мог отследить первые признаки сдавливания мозга вовремя. Если при синдроме Арнольда-Киари болезнь проявляет себя неявно – легкой болью в затылке – врачи назначают консервативное лечение. Самыми популярными при недуге считаются препараты таких групп:
- обезболивающие и противовоспалительные для снятия боли (Ибупофен, Нурофен, Анальгин, Кетопрофен, Нимесулид);
- миорелаксанты для устранения напряжения мышцах (Миорикс, Мускомед, Мидокалм, Сирдалуд);
- мочегонные для уменьшения объема ликвора (Индап, Индапамид, Индапен, Индабрю).
Аномалия Арнольда-Киари – операция
Главная задача при операции – восстановить нормальную циркуляцию ликвора, что впоследствии приведет к улучшению состояния пациента, у которого аномалия Арнольда-Киари 1, 2 или любого другого типа. Во время хирургического вмешательства задняя часть черепной ямки декомпрессируется. Иногда – если болезнь сопровождается гидроцефалией – проблему решают с помощью установки ликворного шунта. По статистике, эффективность операции колеблется в районе от 50 до 85 процентов.
Синдром Арнольда-Киари – осложнения
Это опасная болезнь. Синдром Арнольда-Киари у плода и у взрослого человека может вызывать осложнения, среди которых:
- гидроцефалия;
- застойная пневмония (развивается из-за скопления крови в легких);
- паралич (его вызывает сдавливание спинного мозга, симптомы паралича в самых сложных случаях сохраняются даже после операции);
- нарушения дыхания (иногда даже полная его остановка);
- сирингомиелия (формирование полости или кисты в теле головного или спинного мозга).
Синдром Арнольда-Киари – прогноз
Новости не самые хорошие: при таком диагнозе, как запущенный синдром Арнольда-Киари, выживаемость не слишком велика. Как показывает практика, восстановиться и вернуться к нормальной жизни получается только у пациентов с первыми типами недуга. Чтобы повысить шансы больного, при появлении неврологических симптомов операцию нужно проводить как можно скорее.
Педиатр Анна Колинько о патологии развития головного мозга, которая может встречаться у 30 % населения
Синдром хронической усталости, головокружения и боль в шее могут быть следствием мальформации (аномалии) Арнольда — Киари. После начала широкого использования МРТ стало понятно, что болезнь встречается у 14–30 % популяции
Мальформация Арнольда — Киари (МАК) — это патология развития ромбовидного мозга: продолговатого и заднего мозга, в последний входит Варолиев мост и мозжечок. При МАК задняя черепная ямка не соответствует мозговым структурам, расположенным в этой области: мозжечок и продолговатый мозг из‑за небольших размеров опускаются ниже большого затылочного отверстия, что приводит к их ущемлению и нарушению ликвородинамики. МАК относят к группе кранио-вертебральных (черепно-позвоночных) мальформаций.
В эпоху до МРТ частота МАК оценивалась от 3,3 до 8,2 наблюдений на 100 000 населения, а у новорожденных — 1 на 4–6 тысяч. Сегодня понятно, что распространенность синдрома Арнольда — Киари значительно больше. Из-за бессимптомного течения и в результате учета разных типов МАК цифры очень разнятся — от 14 до 30 %.
Все первые описания мальформации были посмертными. В 1883 году шотландский анатом Джон Клеланд (J. Cleland, 1835–1925 гг.) впервые описал удлинение ствола и опущение миндалин мозжечка в большое затылочное отверстие у 9 умерших новорожденных. В 1891 году австрийский патолог Ганс фон Киари (H. Chiari, 1851–1916 гг.) подробно охарактеризовал 3 типа мальформации у детей и взрослых. А в 1894 году немецкий патолог Юлиус Арнольд (J. Аrnold, 1835–1915 гг.) подробно описал синдром Киари 2 типа, в сочетании со спинномозговой грыжей (spina bifida). В 1896 году Киари дополнил свою классификацию четвертым типом. В 1907 году ученики Арнольда использовали термин «мальформация Арнольда — Киари» по отношению к аномалии 2 типа. Теперь это название распространилось на все типы. Некоторые врачи справедливо отмечают, что вклад Арнольда несколько преувеличен и верным будет термин «мальформация Киари».
Версии о причинах
Этиология и патогенез синдрома Арнольда — Киари остаются неуточненными. Киари предположил, что смещение мозжечка и продолговатого мозга происходит из‑за внутриэмбриональной гидроцефалии, которая возникает как следствие стеноза сильвиева водопровода — узкого канала длиной 2 см, который соединяет III и IV желудочки мозга.
Клеланд полагал, что аномалия связана с первичным недоразвитием ствола головного мозга. В 1938 г. канадский нейрохирург Уайлдер Пенфилд (W. G. Penfield, 1891–1976 гг.) и его коллега предложили «теорию тяги»: в процессе роста фиксированный спинной мозг втягивает в полость позвоночного канала расположенные выше отделы. В «унифицированной» теории Дэвид Маклон (D. G. McLone) и Пол Неппер (P. A. Knepper) в 1989 году предположили, что первично возникает дефект нервной трубки с истечением ликвора и недостаточным расширением желудочковой системы, что приводит к формированию уменьшенной задней черепной ямки. Однако последующие исследования говорят о том, что существуют разные варианты патологии Арнольда — Киари: с уменьшением задней черепной ямки и без такового, с нарушением ликворооттока и без. Описаны семейные случаи МАК 2 типа, однако роль генетических факторов еще недостаточно изучена.
Типы мальформаций
1 тип — опущение миндалин мозжечка в позвоночный канал ниже уровня большого затылочного отверстия с отсутствием спинномозговой грыжи. У 15–20 % пациентов этот тип сочетается с гидроцефалией, а у 50 % больных — с сирингомиелией — заболеванием, при котором в спинном и продолговатом мозге образуются полости. В 1991 году было предложено подразделить аномалии Арнольда — Киари 1 типа на тип А — с сирингомиелией и тип В — без сирингомиелии.
Сирингомиелии при Арнольде — Киари 1 степени.
Энцефаломенингоцеле — врожденная грыжа головного мозга и его оболочек, содержащая цереброспинальную жидкость.
Спинальная дизрафия — порок развития, заключающийся в отсутствии слияния по средней линии парных закладок кожи, мускулатуры, позвонков, спинного мозга
2 тип — опущение нижних отделов червя мозжечка, продолговатого мозга и IV желудочка. Отличительным признаком данного типа является сочетание со спинномозговой грыжей (spina bifida) в поясничном отделе, отмечается прогрессирующая гидроцефалия, часто — стеноз водопровода мозга. Среди детей с менингомиелоцеле до 90 % случаев сопровождается аномалией Арнольда — Киари 2 степени.
0, 1 и 2 степени синдрома Арнольда — Киари наиболее распространены в популяции. III и IV типы обычно несовместимы с жизнью.
Симптоматика
Неврологические симптомы 0 и 1 типов аномалии Арнольда-Киари наиболее часто начинают беспокоить в возрасте 20–40 лет. Степень дислокации миндалин мозжечка может нарастать под влиянием неблагоприятных факторов. Чаще всего жалобами при МАК 0 типа являются головная боль, преимущественно шейно-затылочной локализации, а также боль в шее. Аномалия Арнольда — Киари 1 типа у взрослых чаще проявляется жалобами на нистагм, дизартрию, атаксию, интенционный тремор (тремор при произвольных движениях), головную боль, головокружение, нарушение чувствительности, парезы, нарушение функции тазовых органов, нарушения частоты и ритма пульса, ритма дыхания, лабильность артериального давления, симптомы поражения каудальной группы черепных нервов (IX, X, XI, XII пары) — нарушение чувствительности лица и бульбарные расстройства (расстройства глотания и речи).
Синдром Арнольда-Киари 2 степени впервые проявляется не у взрослых, а у новорожденных или в раннем детском возрасте. МАК 2 типа протекает более тяжело, дети с такой патологией уже рождаются с гидроцефальной формой черепа. Гидроцефалия препятствует нормальному развитию. Кроме того, такие дети страдают нарушениями дыхания, сердцебиения и глотания. Часто заболевание сопровождается судорожными припадками. У детей развивается нистагм, апноэ, стридор, парез голосовых связок, дисфагия с регургитацией, нарушение тонуса в конечностях. Выраженность неврологической симптоматики в первую очередь зависит от выраженности нарушений ликвородинамики, а не от степени эктопии миндалин мозжечка.
Терапия
Лечение аномалий Арнольда — Киари зависит от выраженности неврологической симптоматики. Консервативная терапия включает в себя нестероидные противовоспалительные препараты и миорелаксанты. Если в течение 2–3 месяцев консервативная терапия безрезультатна или у пациента имеется выраженный неврологический дефицит, показано оперативное вмешательство. В процессе операции устраняется сдавление нервных структур и нормализуется ликвороток путем увеличения объема (декомпрессии) задней черепной ямки и установки шунта. Оперативное лечение эффективно, по разным источникам, в 50–85 % случаев, в оставшихся случаях симптоматика регрессирует не полностью. Операцию рекомендуется выполнять до развития тяжелого неврологического дефицита, поскольку восстановление происходит лучше при минимальных изменениях неврологического статуса. Подобное оперативное лечение выполняется почти в каждом федеральном нейрохирургическом центре России и проводится в рамках высокотехнологичной медицинской помощи по системе ОМС.
Пациенты, имеющие мальформацию Арнольда-Киари 0 и 1 типа, могут даже не знать о наличии у себя этого заболевания в течение всей жизни. Вследствие пренатальной диагностики МАК II, III и IV типа дети с данной патологией рождаются всё реже, а современные технологии выхаживания позволяют значительно увеличить продолжительность жизни таких детей.
Синдром Арнольда-Киари – аномалия развития головного мозга, при которой те его отделы, которые расположены в задней ямке черепа, опущены и выдаются через большое затылочное отверстие. Данный синдром может сопровождаться сильными затылочными болями, нистагмом, головокружением, ухудшением зрения и слуха, а также целым рядом других неприятных симптомов. Нарушения, возникающие при аномалии Киари, связаны с работой мозжечка и продолговатого мозга. Как следствие, синдром затрагивает дыхательную систему, ЦНС, крайне негативно отражаясь на работе сердца.
Что такое синдром Арнольда-Киари?

При наличии синдрома наблюдается смещение мозжечка
В месте, где череп соединяется с позвоночным столбом, расположено большое затылочное отверстие. Именно здесь происходит соединение спинного мозга с головным. Чуть выше этого отверстия в черепе присутствует задняя ямка, где располагается мозжечок, мост и продолговатый мозг.
При нормальном развитии головного мозга мозжечок располагается несколько выше затылочного отверстия. При возникновении патологии мозжечковые миндалины опускаются в просвет отверстия, оказывая давление на продолговатый мозг, расположенный под ними. При этом нарушается нормальная циркуляция цереброспинальной жидкости, что запускает процесс развития гидроцефалии.
Нередко патология выявляется у новорожденных. Причиной этому является наличие негативных факторов, влияющих на развитие плода во время беременности. Патология также может быть выявлена и у взрослого человека, причем часто отклонение диагностируется совершенно случайно, никак до этого не проявляясь. В подавляющем большинстве случаев синдром Арнольда-Киари развивается совместно с тяжелым заболеванием нервной системы – сирингомиелией.
Причины развития патологии
В современной медицине нет точных данных относительно этиологии этой аномалии. На данный момент в научных кругах существует несколько гипотез по этому поводу:
- Некоторые специалисты полагают, что данный синдром является следствием недоразвития задней черепной ямки. Если она имеет недостаточные размеры, то структуры, расположенные в ней, по мере роста закономерно опускаются в затылочное отверстие.
- Если головной мозг имеет увеличенные размеры, то это также может стать причиной развития патологии. В таком случае мозжечок, расположенный в черепной ямке, выталкивается из нее мозгом.
- Прогрессирование гидроцефалии. При гидроцефалии наблюдается увеличение мозговых желудочков, а соответственно, и мозг становится больше.
- Вследствие тяжелой черепно-мозговой травмы также может произойти вклинение мозжечковых миндалин в большое затылочное отверстие.
Известен, кроме того, ряд факторов, способствующих развитию синдрома Арнольда-Киари:
- родовые травмы;
- неполноценное питание беременной женщины;
- острые инфекции, перенесенные во время беременности;
- вредные привычки беременной и бесконтрольное употребление ею лекарственных препаратов.
Особенности патологии

Синдром Арнольда-Киари у плода во время беременности
Различают 4 типа болезни Киари:
- Первый тип предполагает незначительное опущение миндалин. Они занимают положение чуть ниже затылочного отверстия. Как правило, данная патология присутствует у подростков и взрослых. Часто сопровождается развитием гидромиелии, когда в центральном спинномозговом канале скапливается большое количество цереброспинальной жидкости.
- Второй тип. Данная форма синдрома Арнольда-Киари развивается у плода ввиду воздействия ряда негативных факторов. Такая патология обычно проявляется у малыша практически сразу же после его рождения. В этом случае через затылочное отверстие проходит не только мозжечок – опускается также продолговатый мозг и IV желудочек. В большинстве случаев одновременно с данной патологией у пациентов выявляется врожденная спинномозговая грыжа.
- Третий тип. При опускании мозжечка и продолговатого мозга они попадают в грыжу головного мозга (менингоцеле), расположенную в шейно-затылочной области.
- Четвертый тип. Не связан со смещением мозжечка, а характеризуется его недоразвитием. Нередко данный тип патологии относят к синдрому Денди-Уокера, характеризующемуся сочетанием гидроцефалии, гипоплазии мозжечка и врожденных кист, локализующихся в задней черепной ямке.
Второй и третий типы аномалии нередко комбинируются с другими проблемами развития ЦНС. Речь идет о гетеротипии коры головного мозга, аномалиях мозолистого тела, гипоплазии подкорковых структур и др.
Симптоматика
Поскольку наиболее распространенным является первый тип аномалии Арнольда-Киари, то его симптоматику следует рассмотреть более подробно. Данный синдром включает в себя несколько симптомокомплексов, а именно:
- церебеллобульбарный;
- ликворногипертензионный;
- сирингомиелитический.
Ликворногипертензионный синдром проявляется следующим образом:
- Сильные головные боли с локализацией в области шеи и затылка. Как правило, они усиливаются при чихании и кашле, а также напряжении шейных мышц.
- Рвота. Позывы возникают вне зависимости от вида питания и периодичности приема пищи.
- Тонус шейных мышц повышен.
- Нарушения мозжечкового характера. К ним относится нистагм, речевые проблемы, мозжечковая атаксия.
Если у пациента присутствует поражение ствола мозга (церебеллобульбарный синдром), то при этом возникают следующие симптомы:
- диплопия (двоение в глазах);
- нарушение глотательной функции;
- ухудшение зрения и слуха;
- шум в ушах;
- сильные головокружения, при которых возникает ощущение того, что окружающие предметы вращаются вокруг пациента;
- потери сознания – кратковременные, но периодически повторяющиеся;
- синдром сонных апноэ;
- парез гортани, сопровождающийся затруднением дыхания и осиплостью голоса;
- атрофия языка (как правило, имеет односторонний характер);
- ортостатический коллапс.
Если данная аномалия развивается параллельно сирингомиелии, то к вышеописанным признакам добавляются следующие симптомы:
- онемение конечностей;
- тазовые нарушения;
- гипотрофия мышц;
- отсутствие брюшных рефлексов;
- нейроартропатия.
Особенности проявления синдрома у детей

Слабый крик, свистящее шумное дыхание у новорожденных – это все симптомы синдрома Арнольда-Киари 2 типа
В детском возрасте проявляется патология второго и третьего типа. Как правило, симптоматика данных отклонений возникает уже в первые дни жизни младенца. Для второго типа синдрома свойственны следующие признаки:
- шумное дыхание;
- двусторонний парез гортани;
- периодическая остановка дыхания;
- нарушение глотательной функции – пища забрасывается в носовую полость;
- нистагм;
- цианоз кожных покровов;
- повышенный тонус мышц в верхних конечностях.
Также возможно появление проблем с передвижением. Такие расстройства могут иметь различные степени тяжести и прогрессировать.
Диагностика
Поставить предварительный диагноз удается уже на основании неврологического осмотра и выслушивания жалоб пациента. Инструментальная диагностика способна показать следующие результаты:
- Эхо-ЭГ, ЭЭГ и РЭГ способны выявить повышение внутричерепного давления, которое нередко возникает при синдроме Арнольда-Киари. Однако посредством этих методик невозможно поставить окончательный диагноз.
- Рентгенография. Такое исследование черепа позволяет определить костные аномалии, следствием которых является развитие синдрома Арнольда-Киари. Ввиду неточности данных эта методика в настоящее время практически не применяется.
- КТ и МСКТ головного мозга. Методы отличаются прекрасной визуализацией костных структур, однако не дают представления о состоянии мягких тканей.
- МРТ головного мозга. Именно это исследование дает наиболее полные результаты относительно состояния мозговых структур, расположенных в области задней черепной ямки.
- МРТ грудного и шейного отделов позвоночника. Позволяет выявить сопутствующие аномалии нервной системы, которые нередко развиваются параллельно с синдромом Арнольда-Киари.
Лечение

Для устранения синдрома Арнольда-Киари применяются обезболивающие и противовоспалительные препараты, а также миорелаксанты
Лечение недуга зависит от степени его проявлений:
- Если синдром протекает бессимптомно и был выявлен случайно, то специфического лечения данной патологии не требуется.
- При наличии у пациента болей в области затылка и шеи применяется консервативная терапия (прием противовоспалительных препаратов, анальгетиков и миорелаксантов).
- При наличии нарушений неврологического характера, которые невозможно устранить консервативно, показано хирургическое вмешательство. Речь идет о проблемах с чувствительностью, парезах, расстройствах мышечного тонуса и т. д.
Особенности хирургического лечения
Для устранения синдрома Арнольда-Киари используются следующие хирургические операции:
- Краниовертебральная декомпрессия. Данная операция предполагает удаление небольшого фрагмента затылочной кости. Это позволяет несколько расширить затылочное отверстие. После этого выполняется резекция мозжечковых миндалин и части двух первых шейных позвонков. Такая процедура позволяет нормализовать циркуляцию церебральной жидкости и устранить симптомы патологии.
- Шунтирование. Операции данного типа предполагают дренирование цереброспинальной жидкости из центрального спинномозгового канала. При этом она может отводиться в брюшную либо в грудную полость.
Прогноз
Прогноз зависит от типа патологии. Аномалия Киари I типа прогноз имеет вполне благоприятный и даже может всю жизнь протекать бессимптомно. III тип синдрома чаще всего быстро заканчивается летальным исходом. В остальных случаях важно своевременное проведение хирургического лечения. Эффективность оперативного вмешательства (краниовертебральной декомпрессии) составляет от 50 до 85%.
